{kind=link}
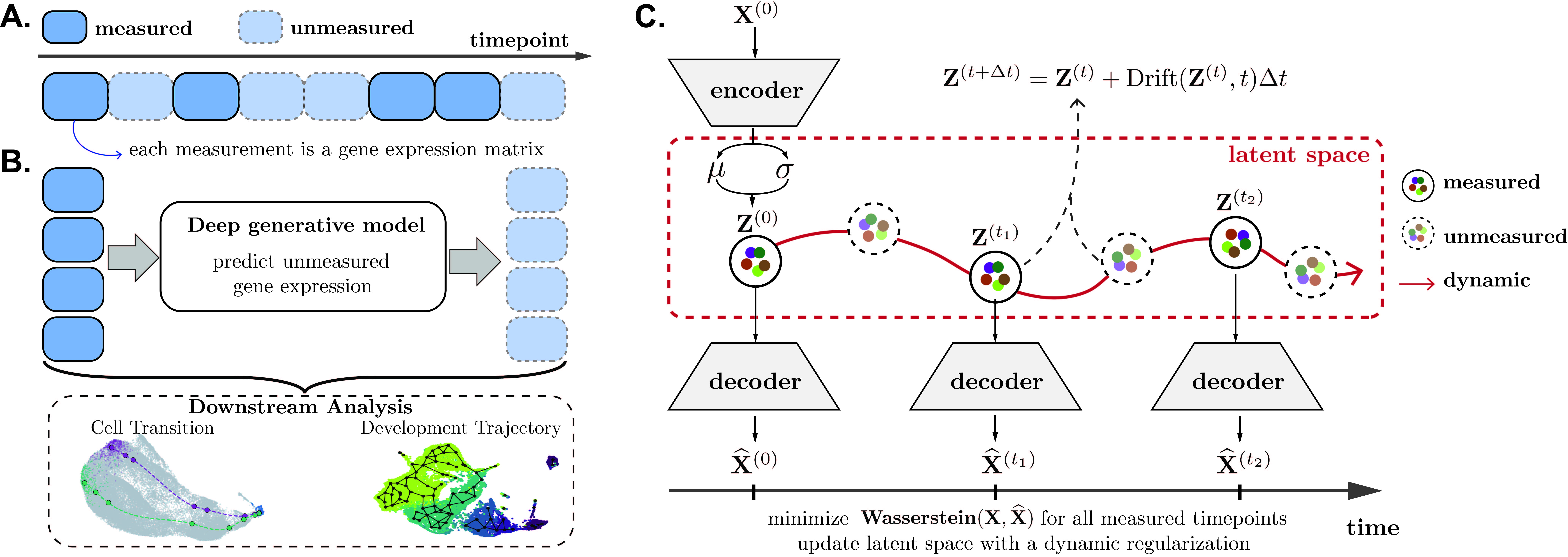
scNODE is a generative model that simulates and predicts realistic in silico single-cell gene expressions at any timepoint. scNODE integrates the idea of variational autoencoder (VAE) and neural ordinary differential equation (ODE) to model cell developmental landscapes on the non-linear manifold. scNODE is scalable to large-scale datasets. (bioRxiv preprint)
If you have questions or find any problems with our codes, feel free to submit issues or send emails to [email protected] or other corresponding authors.
(04/15/2024 updates) We have revised the codes to align with the updated paper.
Our codes have been tested in Python 3.7. Required packages are listed in ./installation.
- Raw and preprocessed data of three scRNA-seq datasets can be downloaded from here.
- All model predictions on three datasets are available at here.
- Experiment results for downstream analysis are available at here.
- Other experimental results, including evaluation metrics, ablation study, and investigation of hyperparameter settings can be downloaded from here.
scNODE is implemented in ./model/dynamic_model.py. We also provide codes of baseline models in ./baseline.
An example of using scNODE is shown in ./benchmark/1_SingleCell_scNODE.py.
- data: Scripts for data preprocessing. Some scripts are implemented in R and need installation of Seurat.
- model: Implementation of scNODE model.
- optim: Loss computations and evaluation metrics.
- baseline: Implementation of baseline models.
- benchmark: Run each model on six scRNA-seq datasets and make predictions. Test scNODE performance with different settings.
- downstream_analysis: Use scNODE for perturbation analysis and help recover smooth trajectories.
- model_validation: Ablation study and investigation of hyperparameter settings.
- plotting: Prediction visualization. Compare model predictions.
- paper_figs: Figure plotting for the bioRxiv preprint.
Please report any bugs, problems, suggestions, or requests as a Github issue